Дистрофия мышц ног лечение. Симптомы мышечной дистрофии, причины и методы лечения
Содержание статьи
Наиболее распространенными среди нервно-мышечных заболеваний являются первичные мышечные дистрофии
. Различные формы миодистрофий отличаются друг от друга типами наследования, сроками начала процесса, характером и быстротой его течения, своеобразием топографии мышечных страданий, наличием или отсутствием псевдогипертрофий и сухожильных ретракций и другими признаками.
Большинство мышечных дистрофий достаточно хорошо изучено клинически, их подробное описание сделано еще в конце прошлого века. Но, несмотря на почти вековую историю изучения миодистрофий, вопросы их патогенеза, достоверной диагностики и лечения остаются до сего времени неразрешенными. Существует большое количество классификаций, но отсутствие точных данных о первичном биохимическом дефекте не дает возможности построить ее по рациональному принципу. В имеющихся классификациях основой являются или клинический принцип, или тип наследования. Так, Walton (1974) предлагает различать следующие формы миодистрофий.
A. Х-сцепленные мышечные дистрофии:
а) тяжелая (тип Дюшенна)
б) благоприятная (тип Беккера)
B. Аутосомно-рецессивные мышечные дистрофии:
а) конечностно-поясная или ювенильная (тип Эрба)
б) детская мышечная дистрофия (псевдодюшенновская)
в) врожденные мышечные дистрофии
C. Лицелопаточно-плечевую (Ландузи - Дежерина)
D. Дистальную мышечную дистрофию
E. Окулярную мышечную дистрофию
F. Окулофарингеальную мышечную дистрофию
Последние несколько форм относятся к аутосомно-доминантным типам наследственной передачи с высокой или неполной пенетрантностью. Следует подчеркнуть, что диагностика мышечных дистрофий нередко представляет большие трудности. Имеется большая вариабельность клинических проявлений, а малое число детей в семье затрудняет определение типа наследования. Наиболее часто встречаются миодистрофии Дюшенна, Эрба и Ландузи - Дежерина.
В настоящее время выявлена значительная группа непрогрессирующих миопатий, которые являются своеобразным пороком развития на уровне мышечной клетки.
Мышечная дистрофия типа Дюшенна
Псевдогипертрофическая мышечная дистрофия Дюшенна - наиболее хорошо изученная форма, встречается чаще других заболеваний мышечной системы (3,3:100 000 населения). Она характеризуется ранним началом и злокачественным течением. Классическая картина проявляется изменением походки у ребенка в возрасте 2-5 лет, к 8-10 годам дети ходят уже с трудом, к 14-15 годам они, как правило, полностью обездвижены. У некоторых детей начальные симптомы проявляются отставанием двигательного развития: они позднее начинают ходить, не умеют бегать и прыгать, при ходьбе отмечается некоторое раскачивание.Одними из первых признаков заболевания являются уплотнение икроножных мышц и постепенное увеличение их объема за счет псевдогипертрофий. Локальные атрофии мышц бедер, тазового пояса нередко маскируются хорошо развитым подкожным жировым слоем. Постепенно процесс принимает восходящее направление и распространяется на плечевой пояс, мышцы спины, а затем и на проксимальные отделы рук. В терминальных стадиях слабость мышц может распространяться на мышцы лица, глотки, дыхательные мышцы.
В развитой стадии болезни имеются такие характерные симптомы, как «утиная походка», подчеркнутый поясничный лордоз, «крыловидные лопатки», симптом «свободных надплечий». Довольно типичны ранние мышечные контрактуры и сухожильные ретракции, особенно ахилловых сухожилий. Рано выпадают коленные рефлексы, а затем рефлексы с верхних конечностей.
Псевдогипертрофии могут развиваться не только в икроножных, но также в ягодичных, дельтовидных мышцах, мышцах живота, языка. Очень часто страдает сердечная мышца по типу кардиомиопатии с изменением ЭКГ на ранних стадиях патологического процесса. При обследовании выявляются нарушение ритма сердечной деятельности, расширение границ сердца, глухость тонов. Острая сердечная слабость - наиболее частая причина летальных исходов при миодистрофии Дюшенна. На вскрытии находят фиброз и жировую инфильтрацию сердечной мышцы.
Довольно характерным симптомом заболевания является снижение интеллекта. Дюшенн, впервые описавший эту форму, обратил внимание на умственную отсталость больных детей. Представляет интерес тот факт, что в одних семьях олигофрения бывает резко выражена, в других - сравнительно умеренно. Изменение высших психических функций не может быть объяснено только педагогической запущенностью больных детей (они рано выключаются из детских коллективов, не посещают детский сад и школу из-за двигательного дефекта). Патоморфологическое исследование после летального исхода выявляет изменения в строении извилин больших полушарий, нарушение цитоархитектоники мозговой коры; ПЭГ у больных показывает развитие гидроцефалии.
Нередко у детей развивается адипозогенитальный синдром, иногда и другие признаки эндокринной недостаточности. Часто находят изменения в костной системе: деформацию стоп, грудной клетки, позвоночника, диффузный остеопороз.
Отличительной особенностью формы Дюшенна, выделяющей ее из остальных мышечных дистрофий, является высокая степень гиперферментемии уже на ранних стадиях развития процесса. Так, уровень специфического для мышечной ткани фермента - креатинфосфокиназы - в сыворотке крови может превышать в десятки и даже сотни раз нормальные показатели. Также значительно повышается активность альдолаз, лактатдегидрогеназы и других ферментов. Лишь в далеко зашедших стадиях болезни степень гиперферментемии постепенно снижается. Имеются сообщения о повышении креатинфосфокиназы на стадии внутриутробного развития. При болезни Дюшенна изменяется обмен креатина. Уже на сравнительно ранних этапах заболевания обнаруживается креатинурия и резко уменьшается экскреция с мочой креатинина. Последний показатель более постоянен и степень уменьшения выделения креатинина в известной степени говорит о тяжести и остроте дистрофического процесса. Наблюдается также увеличение экскреции с мочой аминокислот.
Миодистрофия Дюшенна передается по рецессивному, сцепленному с Х-хромосомой типу. Довольно высока частота мутации гена, что объясняет большое количество спорадических случаев. Для медико-генетического консультирования очень важно установление гетерозиготного носительства. При миодистрофии Дюшенна у заведомых гетерозиготных носителей приблизительно в 70% случаев выявляются субклинические, а иногда и явные признаки мышечной патологии - некоторое уплотнение и даже увеличение икроножных мышц, быстрая утомляемость мышц при интенсивной Физической нагрузке, небольшие изменения на ЭМГ и при патоморфологическом изучении мышечных биоптатов. Наиболее часто у гетерозиготных носительниц выявляется увеличение активности ферментов в сыворотке крови, в частности повышение активности креатинфосфокиназы. Наличие клинических или субклинических признаков заболевания можно объяснить гипотезой Мэри Лайон , согласно которой сумма клеток, содержащих неактивную Х-хромосому с нормальным геном, больше, чем содержащих мутантный ген.
При наличии клинической картины миодистрофии Дюшенна у лиц женского пола следует в первую очередь исключить возможность аномалии по Х-хромосоме - синдром Шерешевского - Тернера (ХО), синдром Морриса (XY) или мозаицизм по этим синдромам.
Мышечная дистрофия типа Беккера-Кинера
Наряду с тяжелой, злокачественной формой Х-сцепленной миодистрофии (тип Дюшенна) существует доброкачественная форма заболевания (тип Беккера - Кинера). По клиническим симптомам она очень напоминает форму Дюшенна, однако начинается, как правило, позднее - в 10-15 лет, течет мягког больные длительно сохраняют работоспособность, в возрасте 20-30 лет и позже еще могут ходить, фертильность не снижена. Заболевание прослеживается в нескольких поколениях семьи, нередко имеется так называемый «эффект деда» - больной мужчина через свою дочь передает заболевание внуку.Впервые доброкачественная форма Х-сцепленной миодистрофии была описана в 1955 г. Becker и Kiner. Начальные симптомы, как и при болезни Дюшенна, проявляются слабостью в мышцах тазового пояса, затем в проксимальных отделах нижних конечностей. У больных изменяется походка, они испытывают затруднение при подъеме по лестнице, при вставании с низкого сиденья. Характерны псевдогипертрофии икроножных мышц. Ретракции ахилловых сухожилий выражены менее резко, чем при болезни Дюшенна. При этой форме не отмечается нарушений интеллекта, почти не встречается кардиомиопатия или она выражена незначительно.
Как и при других Х-сцепленных миодистрофиях, при форме Беккера - Кинера изменяется уровень ферментов в сыворотке крови - значительно повышается активность креатинфосфокиназы, лактатдегидрогеназы и альдолазы, хотя и в меньшей степени, чем при болезни Дюшенна. Нарушается также обмен креатина и аминокислот. В литературе обсуждается вопрос о нозологической самостоятельности болезни Беккера - Кинера. Не решен окончательно вопрос о том, определяются ли формы Беккера - Кинера и Дюшенна различными мутантными аллелями в том же самом генном локусе или в двух различных локусах. McKusick (1962) предполагает, что имеется несколько форм Х-сцепленных мышечных дистрофий, так же как имеется несколько форм цветовой слепоты, гемофилии и дегенерации сетчатки.
Существенным доказательством в пользу нозологической самостоятельности доброкачественной формы заболевания являются некоторые биохимические исследования. Так, было показано, что при миодистрофии Дюшенна происходит высокий коллагеновый и низкий неколлагеновый синтез белка в тяжелых полирибосомах, а при миодистрофии Беккера - Кинера увеличен как коллагеновый, так и неколлагеновый синтез в полисомах. Патоморфологические исследования также выявляют известные различия - при форме Беккера-Кинера имеется отчетливая сохранность регенераторных процессов в мышечной ткани, кроме того, сохранена миоглобинпероксидазная активность в противоположность болезни Дюшенна, где последняя постоянно резко снижена.
При изучении групп сцепления в Х-хромосоме было показано, что локус глюкозо-6-фосфатдегидрогеназы и локус доброкачественной формы Беккера - Кинера находятся ближе, чем локус злокачественной формы Дюшенна. Однако исследования были сделаны только на трех семьях с доброкачественной формой.
Против нозологической самостоятельности этих двух заболеваний говорят накапливающиеся описания семей, в которых имеется сочетание больных с обеими формами. Так,. Walton (1956) описал семью, где 3 брата с болезнью Дюшенна имели 3 дядей по материнской линии, страдающих доброкачественной формой миодистрофии. Furukawa с соавт. (1977) наблюдал 3 семьи, в которых сосуществовали обе формы. Учитывая различное течение и прогноз этих двух форм, рациональнее их оценить как разные заболевания.
Редкие формы Х-сцепленных миодистрофий
В настоящее время известно несколько вариантов сравнительно редко встречающихся наследственных мышечных дистрофий, передающихся через Х-хромосому и (как при форме Беккера-Кинера) имеющих мягкое, благоприятное течение. К таким формам относятся: миодистрофия Дрейфуса - Хогана, форма Мэбри, форма Роттауфа - Мортье - Бейера, формы Робера и Хэка-Лаудана.Форма Дрейфуса-Хогана описана в 1961 г. По срокам начала она напоминает болезнь Дюшенна, чаще это 4-5-летний возраст. Развиваются мышечная слабость и атрофии в мышцах тазового пояса и проксимальных отделов нижних конечностей. Очень медленно процесс распространяется на мышцы плечевого пояса и проксимальных отделов верхних конечностей, иногда вовлекается мускулатура лица, в частности круговая мышца рта. Характерной особенностью этой формы является отсутствие псевдогипертрофий и раннее развитие сухожильных ретракций в ахилловых сухожилиях, л также в сухожилиях двуглавой мышцы плеча и других. Интеллект больного сохранен. Нередко развивается кардиомиопатия с изменением сердечного ритма, чаще всего в возрасте 30-40 лет. Цветовое зрение нормально. Значительно повышается активность ферментов сыворотки крови, особенно возрастает уровень креатинфосфокиназы; в далеко защедших стадиях ферментемия постепенно снижается.
Форма Мэбри известна с 1965 г. Автор и сотрудники наблюдали семью, где в 2 поколениях у 9 лиц мужского пола имелась характерная клиническая картина. Первые симптомы появлялись в пубертатном периоде (11-13 лет) в виде слабости в мышцах бедер и тазового пояса. Имелись выраженные псевдогипертрофии. Для этой формы миодистрофии не характерны сухожильные ретракции, нет цветовой спепоты и других маркеров по Х-хромосомной патологии. Интеллект сохранен. Постоянно страдает мышца сердца. Повышена активность сывороточных ферментов.
При биопсии мышцы выявляются выраженные атрофические изменения с уменьшением размеров мышечных волокон и отсутствием гипертрофированных. Уменьшено количество коллагеновых волокон и резко выражен липоматоз.
Форма Роттауфа - Мортье - Бейера описана впервые в 1971 г. Авторы наблюдали большую семью, где в 4 поколениях было 17 больных мужчин. Характерной особенностью этой формы является развитие ранних и резко выраженных сухожильных ретракций и мышечных контрактур. Эти симптомы появляются в возрасте 5-10 лет вначале в дистальных отделах ног (ограничение тыльного сгибания стоп), затем развивается ограничение сгибания шеи и разгибания в локтевых суставах. Постепенно формируются патологические позы головы и туловища из-за прогрессирующего фиброза мышц с невозможностью сгибания позвоночника. Парезы выражены очень умеренно, в основном в мышцах плечевого пояса, а также в дистальных отделах ног; мышечные гипотрофии диффузные, но нерезкие. Псевдогипертрофии полностью отсутствуют.
Интеллект у больных сохранен (среди них встречаются даже одаренные люди). Грубо страдает сердечная мышца, как правило, постепенно формируется нарушение проводимости и к 35-40 годам может развиться полная атриовентрикулярная блокада. ЭМГ и данные биопсии указывают на миогенный характер изменений. Отмечается четкая гиперферментемия, степень которой снижается в далеко зашедших стадиях процесса. У гетерозиготных носительниц нет клинических проявлений и показатели активности ферментов нормальны.
Прогрессирование заболевания очень медленное, больные длительно сохраняют возможность самообслуживания и даже трудоспособность. Многие вступают в брак и могут иметь детей. Фертильность не ограничена. Летальный исход, как правило, наступает в возрасте 40-50 лет и обусловлен поражением сердечной мышцы.
Конечностно-поясная форма мышечной дистрофии (ювенильная миопатия Эрба)
Встречается с частотой 1,5:100 000 населения. Передается по аутосомно-рецессивному типу наследования, оба пола страдают одинаково часто.Начало заболевания в большинстве случаев относится к середине второго десятилетия жизни (14-16 лет), однако имеется довольно широкий возрастной диапазон. Описана так называемая ранняя, или псевдодюшеновская, форма, когда первые симптомы проявляются в возрасте до 10 лет и течение заболевания более тяжелое. Имеется и поздний вариант с началом после 30 лет.
Течение заболевания может быть быстрым или более медленным, в среднем полная инвалидность наступает через 15-20 лет от начала появления первых симптомов. В большинстве случаев миодистрофия Эрба начинается с поражения мышц тазового пояса и проксимальных отделов ног, где появляются слабость и похудание мышц. Процесс в дальнейшем распространяется на плечевой пояс. В некоторых случаях плечевой п тазовый пояса поражаются одновременно. Довольно значительно страдают мышцы спины и живота. У больных имеется характерная «утиная» походка, затруднено вставание из положения лежа и сидя, подчеркнут поясничный лордоз. Мышцы лица в большинстве случаев не страдают. Для этой формы миодистрофии сравнительно мало характерны значительные контрактуры и псевдогипертрофии. Могут иметь место концевые атрофии и сухожильные ретракции. Интеллект у больных обычно сохранен. Сердечная мышца большей частью не поражена. Уровень ферментов в сыворотке крови, как правило, повышен, однако далеко не столь резко, как при Х-сцепленной миодистрофии. Есть указания, что у больных мужского пола уровень креатинфосфокиназы выше, чем у больных женщин. Имеется значительная разница в экспрессивности мутантного гена у разных членов семьи - наряду с тяжелой клинической картиной могут быть сравнительно легкие и даже стертые клинические симптомы. Нарушается креатин-креатининовый обмен, особенно резко уменьшается экскреция креатинина, увеличено выделение альфааминоазота с мочой. На ЭМГ находят изменения миогенного типа со снижением амплитуды биопотенциалов и сохранной частотой.
Миодистрофия Эрба - наиболее аморфная форма и большинство фенокопий имитирует именно эту форму патологии, поэтому очень важно в спорадических случаях проводить тщательное клиническое обследование для исключения в первую очередь воспалительного поражения мышц типа полимиозита, особенно при наличии болевого синдрома, а также эндокринных миопатий, токсических, лекарственных, карциноматозных и других миопатий. Подобные фенокопии встречаются особенно часто в пожилом возрасте.
Лицелопаточно-плечевая форма миодистрофии (тип Ландузи-Дежерина)
Эта форма миодистрофии описана в 1884 г. Landousi и Dejerine. Встречается реже, чем две предыдущие формы (0,9: 100ООО населения). Заболевание передается по регулярному аутосомно-доминантному типу с высокой пенетрантностью и несколько вариабельной экспрессивностью. По данным некоторых авторов, женщины болеют чаще мужчин (3: 1). Физические перегрузки, интенсивные занятия спортом, а также нерационально проводимая лечебная физкультура могут способствовать более тяжелому течению болезни.Миодистрофия Ландузи--Дежерина - сравнительно благоприятно текущая форма мышечной патологии. Начинается она чаще в возрасте около 20 лет, иногда позже. Однако в семейных случаях заболевания, когда можно проследить за младшими членами семьи в динамике удается выявить некоторую слабость мышц, например лица, в более раннем возрасте.
По-видимому, вначале слабо выраженные симптомы длительное время остаются стабильными, а затем наступает нрогредиентность течения. Больные доживают до солидного иозраста (60 лет и более).
Мышечная слабость и атрофии вначале появляются в мышцах лица или плечевого пояса. Постепенно эти нарушения распространяются на мышцы проксимальных отделов рук, а затем и на нижние конечности. Характерно, что в большинстве случаев вначале поражаются мышцы передней поверхности голеней, затем мышцы проксимальных отделов ног. На высоте заболевания грубо страдают круговые мышцы глаза н рта, большая грудная, передняя зубчатая и нижние отделы трапециевидной мышц, широкая мышца спины, двуглавая, трехглавая мышцы плеча. Характерен внешний вид таких -больных: типичное лицо «миопата» с «поперечной улыбкой», резко выраженные «крыловидные лопатки», своеобразная деформация грудной клетки за счет мышечного скелета с уплощением ее в переднезаднем направлении и ротацией внутрь плечевых суставов. Нередко имеется асимметрия поражения, даже в пределах одной мышцы (например круговой мышцы рта). Наблюдаются псевдогипертрофии икроножных, дельтовидных мышц, иногда мышц лица. Контрактуры и ретракции выражены умеренно. Сухожильные рефлексы длительное время бывают сохранены.
Признаки поражения сердечной мышцы выявляются редко и они практически не отличаются от таковых в общей популяции, хотя нарушения атриовентрикулярной проводимости описаны. Уровень активности сывороточных ферментов увеличен незначительно, может быть даже нормальным. Креатин-креатининовый обмен нарушен умеренно, хотя некоторое уменьшение креатинина в моче выявляется постоянно. Интеллект у больных при этой форме не страдает. Представляет интерес тот факт, что ЭМГ у больных миодистрофией Ландузи - Дежерина нередко не совсем типична для мышечного уровня поражения. У некоторых больных (членов одной семьи) может наблюдаться характерное снижение амплитуды биопотенциалов, интерференционный тип кривой, у других, наоборот, уменьшение частоты и гиперсинхронная активность, иногда с типичным ритмом частокола. Следует помнить о наличии неврогенного варианта плечелопаточно-лицевой миодистрофии.
В настоящее время ряд авторов полагают, что форма Ландузи-Дежерина не является единой, гомогенной формой, а представляет собой синдром. Лицелопаточно-плечевой синдром встречается при миодистрофии Ландузи - Дежерина, неврогенной амиотрофии, при миастении, миотубулярной, немалиновой миопатии, митохондриальной миопатии и центроядерной миопатии. Клинический диагноз должен подкрепляться, помимо электромиографического исследования, результатами гистохимического и электронно-микроскопического исследований.
Дистальная форма мышечной дистрофии
Первое сообщение об этой форме мышечного поражения относится к 1907 г. Spiller привел клинические и патологоанатомические данные и отметил, что заболевание отличается от невральной амиотрофии Шарко - Мари. Подробное клиническое описание дистальной формы мышечной дистрофии дано в 1951 г. Welander, которая наблюдала боле 250 больных в Швеции. Заболевание встречается сравнительно редко. Тип наследования - аутосомно-доминантный с неполной пенетрантностью и вариабельной экспрессивностью.Первые симптомы заболевания проявляются в сравнительно позднем возрасте, как правило, позже 20 лет, хотя имеются описания болезни с началом в 5-15 лет . Заболевание отличается доброкачественным течением. Поражаются дистальные отделы нижних конечностей - появляются парезы стоп, голеней, развивается мышечное похудание. Постепенно слабость и гипотрофии распространяются на кисти и предплечья, в далеко зашедших случаях могут страдать проксимальные отделы ног. Выпадают сначала ахилловы рефлексы, затем коленные и рефлексы на руках. Не наблюдается псевдогипертрофий, фасцикуляций, всегда сохранена чувствительность. Сухожильные ретракции также мало характерны. В очень редких случаях развивается кардиомиопатия.Заболевание не всегда легко разграничить с невральной амиотрофией Шарко - Мари. Опорными пунктами в диагностике служат данные электрофизиологических методов исследования. При дистальной миопатии всегда нормальна скорость проведения возбуждения по нервному стволу, ЭМГ указывает на мышечный тип поражения. Следует отметить, что наблюдается неврогенная амиотрофия с локализацией парезов и похуданий мышц в дистальных отделах рук и ног. В этих случаях ЭМГ регистрирует типичный спинальный характер биоэлектрической активности с урежением частоты и явлениями синхронизации. Важным диагностическим критерием является исследование сывороточных ферментов, активность которых может значительно увеличиваться при мышечной дистрофии и не меняться при спинальной и невральной амиотрофии. Четкая креатинурия и резкое снижение экскреции с мочой креатинина будут свидетельствовать также в пользу миогенной природы страдания.
Окулярная и окулофарингеальная миопатии
Изолированное первичное поражение мышц глазного яблока впервые отмечено Говерсом и Мебиусом около 100 лет тому назад, однако подробное описание этой формы поражения дано в 1951 г. Kilon. Заболевание встречается редко. Тип наследственной передачи аутосомно-доминантный, с низкой пенетрантностью. Часто имеют место спорадические случаи.Начало заболевания в возрасте 25-30 лет, но иногда первые симптомы отмечают в пубертатном периоде. Вначале появляются небольшой птоз, который постепенно увеличивается, затем ограничение движений глазного яблока, как правило, симметричное. Жалобы на двоение в глазах крайне редки. Течение заболевания медленно прогрессирующее, обычно до полной наружной офтальмоплегии. Внутренние мышцы глаза не страдают. Процесс на этом иногда приостанавливается, однако в ряде случаев присоединяется слабость круговой мышцы глаза, лобной мышцы и других мимических мышц. На ЭМГ и при исследовании биоптата обнаруживается заинтересованность мышц шеи и плечевого пояса; иногда парезы и гипотрофия этих мышц выявляются и клинически. В редких случаях отмечается широкая генерализация процесса.
При окулофарингеальной миопатии, которая встречается еще реже, в процесс включаются также мышцы глотки и мягкого неба. Это заболевание встречается после 40 лет. В подобных случаях, помимо офтальмоплегии, развивается дисфагия и дисфония.
При патоморфологическом исследовании выявляются разнокалиберность мышечных волокон, наличие маленьких ангулярных волокон, вакуолярные изменения. Разрастание соединительной ткани, фагоцитоз и базофилия встречаются нечасто. Во многих случаях находят измененные митохондрии, которые нередко увеличены в размере, кристы в них располагаются неправильно - по периферии.
Дифференциальный диагноз в ряде случаев труден с особой формой глазной миастении. Этой формой миастении болеют чаще лица мужского пола, начало ее нередко острое, возраст больных от 20 до 30 лет. Характерно течение без ремиссий, имеет место резистентность к антихолинэстеразным препаратам. Решающим в диагностике является электромиографическое исследование с ритмической стимуляцией и проведением тестов с кураре или тензилоном.
Проводят также дифференциальный диагноз с органическими поражениями головного мозга (опухоли среднего мозга, воспалительные процессы мозга и его оболочек).
Редкие формы прогрессирующих мышечных дистрофий. Описано значительное число больных с врожденной мышечной дистрофией, у которых имелась картина «floppy baby» . У части таких больных диффузная мышечная слабость, гипотония, обнаруживаемые при рождении, могут сочетаться с множественными контрактурами (тип артрогрипоза). Дети с подобными формами рано умирают. Тип наследования аутосомно-рецессивный.
К редким формам мышечных дистрофий относится миопатия четырехглавых мышц бедра и ряд других миопатий.
Патоморфологнческие изменения при прогрессирующих мышечных дистрофиях
Изменения со стороны нервной системы при мышечных дистрофиях отсутствуют или они минимальны. Описана патология спинного мозга, в котором иногда находят уменьшение клеток передних рогов. Отмечают изменения двигательных нервных окончаний (осевых цилиндров и миелиновых оболочек).Отмечено нарушение структуры моторных бляшек с исчезновением фибриллярной структуры .
Основные изменения отмечены в самой мышечной ткани. Мышечные волокна истончаются, замещаются жировой и соединительной тканью, отдельные волокна гипертрофируются, увеличивается количество мышечных ядер. Последние могут располагаться цепочками. Находят изменения в сосудах - утолщение стенок, стенозирование, иногда наблюдаются микротромбозы. Гистохимическое исследование мышечного биоптата позволяет выявить накопление кислых мукополисахаридов, уменьшение ряда ферментов. При электронно-микроскопическом изучении было установлено разрушение миофиламентов, расширение межфибриллярных пространств, изменение z-полос, увеличение каналов саркоплазматического ретикулума с образованием вакуолей. Изменяется структура митохондрий, они могут приобретать шаровидную форму, атрофируются кристы, как правило, увеличивается число лизосом.
Патогенез прогрессирующих мышечных дистрофий
Вопросам изучения патогенеза прогрессирующих мышечных дистрофий посвящено огромное количество исследований, однако до сего времени первичный биохимический дефект не открыт, не выяснены механизмы гибели мышечных волокон, неизвестны причины избирательного поражения мышц при различных формах миодистрофии. На современном этапе не утратили своего значения следующие гипотезы: неврогенная, гипоксическая, дефектных мембран, дисфункции внутриклеточных медиаторов.Неврогенная гипотеза предполагает первичное поражение нервной системы (спинного мозга, а также периферических ее отделов, включая внутримышечные волокна) с вторичным нарушением метаболизма мышечной ткани. В основе этой гипотезы лежат данные, указывающие на уменьшение мотонейронов в передних рогах спинного мозга, наличие дегенеративных изменений в нервных терминалях и концевых пластинках, уменьшение количества моторных единиц в дистрофических мышцах, изменение аксонального тока структурных белков и низкомолекулярных соединений, некоторое замедление проводимости возбуждения в дистальных отделах нервных стволов. Эти данные являются новым подкреплением старых представлений о патогенезе мышечных дистрофий как следствие нарушения трофической функции нервной системы, в частности ее симпатического отдела.
Особый интерес представляет концепция поломки бета-адренорецепторов, что может привести к снижению чувствительности к действию медиаторов вегетативной системы - адреналину и норадреналину и к автономии обмена в мышечной ткани [Хохлов А. П., 1977; Mawatary, 1975]. Несмотря на кажущуюся стройность неврогенной гипотезы, первичность выявленных изменений полностью не доказана. Так, компьютерный метод подсчета моторных единиц не установил существенных различий между нормальной и дистрофичной мышцами. Исследование передних рогов спинного мозга при вскрытии умерших от миодистрофии Дюшенна не выявило патологии. Изменение нервных терминалей, а также аксонального тока веществ может быть вторичным как результат грубого дистрофического процесса в мышце с восходящим перерождением нервных волокон. С позиций неврогенной гипотезы нельзя объяснить клинические и биохимические особенности разных форм миодистрофий. Однако все это не исключает участия нервной системы в общем комплексе патогенетических механизмов.
Гипотеза тканевой гипоксии объясняет гибель мышечных волокон как следствие хронического недостатка кислорода. Предпосылками для этой гипотезы явились патоморфологические данные о сходстве изменений мышц у животных с экспериментальной гипоксией и у больных с миодистрофиями, создание экспериментальной модели миопатии с помощью искусственной эмболизации частицами декстрана, а также повторными инъекциями смеси имипрамина и серотонина. Увеличение кислых мукополисахарндов в основном веществе мышц и стенках сосудов с прогрессирующим новообразованием коллагеновых волокон, а затем формированием плотного фиброзного футляра вокруг мышечных волокон, последующим сдавленней сосудов, хроническим нарушением микроциркуляции (Ситников В. Ф., 1973, 1976] на ранних стадиях миодистрофического процесса служит известным доказательством данной гипотезы. Изучение обмена миоглобина, показавшее его дефектность (близость к фетальному, т. е. функционально неполноценному), еще больше подкрепило гипотезу тканевой гипоксии как первопричины развивающейся гибели мышечной ткани.
Однако последующие контрольные исследования с применением более современных методов не смогли полностью подтвердить эту гипотезу. Так, измерение мышечного кровотока с помощью радиоактивного ксенона выявило нормальный уровень. Электронно-микроскопическое исследование не подтвердило наличия сосудистой окклюзии, а морфометрический анализ капилляров показал их нормальное количество. Повторные эксперименты с эмболизацией сосудов суспензией сухих частиц декстрана не дали возможности получить модель, характерную для миопатии.
Не находит объяснения с позиций этой гипотезы механизм гибели белых мышечных волокон, хотя источником энергии в них является анаэробный гликогенолиз. Не было также получено доказательств разобщения тканевого дыхания и окислительного фосфорилирования.
Гипотеза дефектных мембран. Согласно этой гипотезе, первичным в патогенезе мышечных дистрофий является увеличение проницаемости сарколеммы, а также субклеточных мембран - лизосомальных, митохондриальных, саркотубулярных, в связи с чем происходит потеря таких веществ, как внутриклеточные ферменты, гликоген, аминокислоты, креатин и т. д. Все это приводит к уменьшению количества жизненно важных белков, нарушению баланса биохимических процессов. Обнаружение подобных сдвигов у гетерозиготных носителей подтверждает первичный характер этих структурных нарушений. Однако ряд фактов не находит объяснения с позиций данной гипотезы. Так, было показано, что нарушение проницаемости мембран имеет избирательный характер - такие вещества, как миоглобин, карнитин не выходят из мышечной клетки. Проницаемость мембран существенно меняется при некоторых фармакологических нагрузках, гормональных воздействиях. Трудно объяснить максимальный уровень гиперферментемии при болезни Дюшенна в доклинической стадии при отсутствии еще некроза мышечной ткани, а также механизм гибели мышечных волокон при доброкачественных формах миодистрофии, где степень ферментемии и креатинурии очень незначительна.
Гипотеза нарушения обмена циклических нуклеотидов. Циклические нуклеотиды (циклический аденозинмонофосфат -ц. АМФ, циклический гуанинмонофосфат - ц. ГМФ) играют ведущую роль в процессах метаболизма мышечного волокна, ц. АМФ контролирует активность ключевых ферментов углеводного и липидного обмена, кальцийсвязывающую способность саркоплазматического ретикулума, работу генетического и белково-синтетического аппарата, проницаемость сарколеммы и лизосомальных мембран. Свое регулирующее влияние на метаболизм внутри клетки п. АМФ осуществляет через систему протеинкиназ. Основные биохимические признаки дистрофического процесса (эмбриональные черты метаболизма, накопление жира, усиление протеолиза, переход веществ в кровяное русло) таким образом могут быть объяснены нарушением обмена циклических нуклеотидов.
Уровень ц. АМФ зависит от активности его ферментов- встроенной в мембрану (связанную с бета-адренорецепторами) аденилатциклазы, катализирующей синтез, и фосфодиэстеразы, осуществляющей распад нуклеотида до неактивного АМФ. Содержание ц. АМФ можно изменить, вводя ингибиторы и активаторы этих ферментов. Так, метилксантины, цитрат натрия, тормозя активность фосфодиэстеразы, увеличивают концентрацию нуклеотида. Тот же эффект может быть получен введением адреналина и фторида натрия, стимулирующих аденилатциклазу.
Бета-адреноблокаторы (пропранолол) и активаторы фосфодиэстеразы (имидазол) снижают уровень ц. АМФ.
Немногочисленные данные, имеющиеся в литературе, говорят о том, что у больных миодистрофией Дюшенна ослаблены реакции, находящиеся под контролем циклических нуклеотидов.
Искусственное увеличение уровня ц. АМФ при назначении больным с болезнью Дюшенна метилксантинов в субмаксимальных суточных дозах уже через несколько часов приводит к резкому снижению ферментемии, креатинурии и аминоацидурии, а также улучшению общего состояния больного. Дополнительная блокада бета-адренорецепторов (например, при введении пропранолола) у этих больных дает обратные биохимические сдвиги и приводит к ухудшению самочувствия, увеличению мышечной слабости.
У больных с миодистрофией Эрба и Ландузи-Дежерина установлен противоположный характер метаболических сдвигов по сравнению с Х-сцепленными формами миодистрофии. Так, 10-дневный курс лечения анаприлином приводит к закономерному уменьшению креатинурии в среднем на 40%, аминоацидурии на 50%, активности КФК более чем в 1.5 раза[Полякова Н. Ф„ 1978].
Полученные данные о важной роли ц. АМФ в развитии дистрофического процесса показали таким образом различный характер биохимических сдвигов при разных формах мышечных дистрофий. Они послужили основой для разработки принципиально нового метода лечения миодистрофии Эрба и Ландузи - Дежерина с применением бета-адреноблокаторов. Остается недостаточно доказанной первичность выявленных изменений обмена циклических нуклеотидов.
Лечение при первичных мышечных дистрофиях
Отсутствие данных о первичном биохимическом дефекте и патогенезе заболевания затрудняет проведение рациональной терапии.Накопленный опыт говорит о том, что систематическое проведение комплексных курсов лечения в ряде случаев способствует некоторому замедлению патологического процесса, иногда и его стабилизации.
Во все комплексы должны включаться ЛФК и массаж, которые способствуют поддержанию мышечного тонуса, улучшают периферическое кровоснабжение, задерживают развитие контрактур. Важное место отводится дыхательной гимнастике. Аналогичный принцип лежит в основе рекомендаций назначения сосудорасширяющих препаратов в сочетании с оксигенотерапией, физиотерапией, бальнеотерапией (радоновые или сульфидные ванны). Следует иметь в виду, что физиотерапия и особенно бальнеотерапия рекомендуются только в ранних стадиях процесса или при доброкачественных, медленно прогрессирующих формах миодистрофий.
Назначение анаболических гормонов должно проводиться с большой осторожностью, короткими курсами (ретаболил 1 раз в 5-7 дней, на курс лечения 5-6 инъекций) с одновременным назначением переливания одногруппной крови по 100-150 мл. Прямым показанием для введения этой группы препаратов является гипогонадизм у лиц мужского пола.
Могут быть рекомендованы витамин Е внутрь или внутримышечно (инъекции эревита), витамины группы В, никотиновая кислота. Показано лечение монокальциевой солью АТФ по 3-6 мл в день внутримышечно в течение месяца.
Проводят лечение аминокислотами (гликокол, лейцин, глутаминовая кислота) и оротатом калия.
В современной неврологии существует огромное количество заболеваний, характер возникновений которых не поддаются рациональному объяснению со стороны специалистов. К таким болезням можно отнести группу таких недугов, как мышечная дистрофия. Всего разновидностей данной болезни девять, но обо всем по порядку…
Мышечная дистрофия - это хроническое наследственное заболевание в результате развития которого происходит поражение мышечной системы человека. Пораженные мышцы перестают нормально функционировать, истончаются в размерах, а в месте их расположения в организме постепенно нарастает жировая прослойка.
Разновидности мышечной дистрофии
В современной неврологии данный недуг классифицируется на девять различных болезней. Классификация заболевания связана с:
- локализацией мышечных нарушений;
- характеристиками болезни;
- агрессивностью развития;
- возрастом.
Так, мышечная дистрофия бывает:
- Дюшенна;
- миотоническая (болезнь Штейнерта);
- Беккера;
- Эрба Рота;
- юношеская форма дистрофии Эрба-Рота;
- плече - лопаточная лицевая форма (Ландузи-Дежерина);
- алкогольная миопатия;
- дистальная форма;
- миодистрофия Эмери -Дрейфуса.
Дистрофия Дюшенна
Наиболее известная форма прогрессирующая дистрофия Дюшенна (псевдогипертрофическая дистрофия, мерозин - негативная врожденная дистрофия). Данная разновидность недуга проявляется в детском возрасте начиная с двух до пяти лет. В первую очередь от данного недуга страдают мышцы нижних конечностей, которые несмотря на малоподвижный образ жизни у маленьких пациентов, постепенно увеличиваются в размерах. Данная особенность связана с нарастанием жировой ткани вместо мышц.

Слева здоровый ребенок, справа больной
Постепенно, по мере прогрессирования болезни она переходит в верхнюю часть и поражает мышцы верхних конечностей. Как правило, уже к двенадцатилетнему возрасту маленький пациент перестает полностью двигаться. Летальность у данного заболевания очень высока примерно 85–90% больных не доживают до 20-летнего возраста.
В группе риска находятся представители мужского пола, так как девочек данный недуг практически не затрагивает.
Болезнь Штейнерта
Врожденная дистрофия Штейнера не зря имеет название миотоническая, так как в результате прогрессирования болезни происходит слишком медленное расслабление мышц после их сокращения (такое явление называется миотонией).
Данное заболевание, в отличие от предыдущего распространено у взрослых, в возрасте от 20 до 40 лет. Имеют место быть и случаи прогрессирования болезни у детей, как правило, в младенческом возрасте, однако, это скорее исключение, чем правило.

На лицо признаки миотонии (приоткрытый рот и веки)
Гендерной зависимости болезнь не имеет и в равной степени от нее страдают как мужчины, так и женщины. Отмечается, что при недуге проявляется слабость мимических мышц лица, а также конечностей. Прогрессирование, в отличие от болезни Дюшенна происходит медленно.
Отличительная черта заболевания, возможность поражения не только мышц конечностей, но и внутренних мышц (сердечная мышца), что в свою очередь, представляет большую опасностью для жизни человека.
Болезнь Беккера
Данная форма болезни также долго прогрессирует, и пациент длительное время чувствует себя хорошо. Обострение недуга может произойти на фоне полученных травм или различных заболеваний нервной системы, которые своим течением ускорят развитие недуга.
В группе риска находятся люди низкого роста.
Болезнь Эрба-Рота и юношеская ее форма
Данное аутосомно - рецессивное заболевание развивается у пациентов после 20 лет, а юношеская форма у детей и подростков 11-13-летнего возраста. Прогрессирование болезни происходит по восходящему варианту, то есть сначала поражаются мышцы нижних конечностей, и постепенно происходит восхождение недуга к верхним конечностям.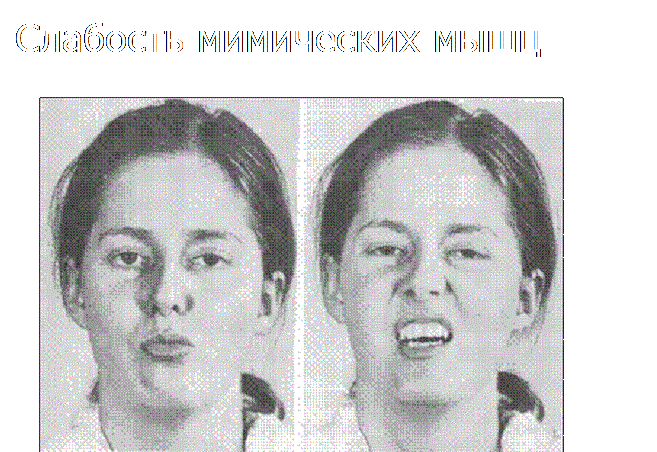
Отличительной чертой недуга является наличие выступающих лопаток, которые по мере прогрессирования выступают более явно и очевидно.
Во время ходьбы отмечается переваливание пациента, выпячивание живота и задвигание грудной клетки.
Болезнь Ландузи-Дежерина
Плече-лопаточно-лицевая форма данного заболевания является наиболее безразборным видом, так как поражает людей в возрасте от пяти до 55 лет. Для данного недуга характерно очень длительное развитие, пациент может сохранять трудоспособность до 25 лет жизни с данной болезнью.
Отличительными симптомами является поражение лицевых мышц, в результате чего у больного могут возникнуть проблемы с четкостью произношения, в связи с неполным смыканием губ. Кроме того, отмечается неполное смыкание век.
По мере развития у больного атрофируются лицевые мышцы, мышцы плеч, конечности и туловища.
Данная форма не зависит от генетической мутации.
Алкогольная миопатия
Данный вид болезни также несвязан с генными мутациями и причина его возникновения одна - злоупотребление алкоголем. У больных могут отмечаться болевые синдромы в конечностях, связанные именно с поражением мышц.
Различают острую и хроническую алкогольную миопатию.
Дистальная форма
Дистальная форма мышечной дистрофии является доброкачественным вариантом прогрессирующей дистрофии. Как правило, данное заболевание трудно дифференцировать от невральной амиатрофии Мари-Шарко. Для разделения этих двух заболеваний требуется проведение электроэнцефалограмму головы, которая и дает понимание с какой болезнью предстоит иметь дело.
К основным симптомам недуга относят атрофию мышц конечностей с их последующим исхуданием. Возможны парезы стоп, кистей рук и т. п.
Миодистрофия Эмери -Дрейфуса
Данный тип болезни первоначально, вообще, не был выделен в качестве отдельного заболевания, так как по своей симптоматике был схож с дистрофией Дюшенна. Однако, в дальнейшем в результате длительного исследования было установлено, что болезнь Эмери -Дрейфуса обладает индивидуальной симптоматикой.
Болезнь относят в разряд редких. В группе риска находятся люди до 30 лет, как правило, юный возраст. Есть свидетельства о проявлении симптомов и после 30 лет, но они единичны.
Основное отличие данного вида болезни - это проблемы с мышцами сердца, которые в итоге могут стать причиной смерти. Кардиомиопатия при данном недуге не единственное отличие. Кроме проблем с сердцем, у пациентов наблюдают стандартные для дистрофии Дюшенна признаки, но в более щадящем режиме развития.
Причины
Негативная составляющая болезней нервной системы заключается в том, что они тяжело поддаются изучению. По этой причине не до конца известно о факторах, провоцирующих развитие той или иной формы мышечной дистрофии.
Основная причина, для формирования большинства подвидов данной болезни является генная мутация, в частности гена, который отвечает за синтез белка.
К примеру, болезнь Дюшенна имеет прямое отношение к мутации половой Х хромосомы. Основной переносчик нехорошего гена выступают девушки, которые несмотря на его наличие в собственной ДНК не страдают от данной болезни.
Что касается миотонической формы, то виновник ее возникновения ген, расположенный в 19 хромосоме.
Основные симптомы
Наличие большого количество различных подвидов у данного недуга указывает на различия в симптоматике, однако у болезни есть общие симптомы, которые в себя включают:

Диагностика мышечной дистрофии
Диагностические мероприятия при подобном недуге обширны, так как существует большое количество болезней, от которых необходимо дифференцировать болезнь.
На первоначальном этапе доктор обязательно изучит анамнез больного, уточнит о симптоматике, режиме дня и т. п. Эти данные требуются для составления плана проведения последующих диагностических мер, которые могут в себя включать:

Стоит отметить, что чем позже проявилась болезнь, тем лучше для больного, так как ранние симптомы в большинстве случаев заканчиваются смертью.
Лечение
Лечение мышечной дистрофии процесс сложный и затяжной, однако, в настоящее время не создано лекарство, которое полностью исцеляет больного. Все мероприятия направлены на облегчение жизни больного и восстановления некоторых утраченных способностей.
Для затормаживания развития болезни назначают следующие препараты:
- кортикостероиды;
- витамины В1;
- аденозинтрифосфат (АТФ).
Кроме того, для замедления процесса развития используют фетальные стволовые клетки, которые замедляют процесс дистрофии.
Помимо того, в качестве профилактических мероприятий назначают:
- массаж;
- физиотерапию;
- дыхательную гимнастику.
Помимо стандартных вариантов лечения, важно постоянно руководствоваться тремя главными составляющими в процессе жизнедеятельности:
- Адекватная физическая активность.
- Своевременная психологическая поддержка.
- Соблюдение диеты.
Физическая активность
Отсутствие желания у человека бороться с недугом оказывает негативное воздействие на организм. Судите сами, пассивность, нежелание двигаться угнетает и без того пораженную мышечную систему. Мышцам необходимо давать нагрузку, так как без нагрузки дистрофичные процессы начинают происходить быстрее, тем самым прогрессируя в более быстром темпе.
Умеренная физическая активность, использование поддерживающих приспособлений будут отличным подспорьем в борьбе с недугом.
При наличии болей в мышцах отлично подойдет плавание, йога, упражнения на растяжку.
Психологическая поддержка
Психологическая поддержка окружения важна для больного человека. А если недуг такой серьезный, как этот тем более. Для кого-то будет достаточно обычной поддержки со стороны друзей и родных, а кому-то может потребоваться квалифицированная психологическая помощь.
Важно дать понять такому человеку, что он не остался один на один со своей проблемой. Он должен понимать, что ему есть к кому обратиться, есть люди, которые сопереживают и поддерживают его.
Диета
Что касается режима питания и соблюдения диеты, существует расхожее мнение, что соблюдение противовоспалительной диеты может замедлить прогрессирование недуга. Данная диета снижает воспаление, уровень глюкозы, выводит токсины из организма и питает его полезными веществами.
Суть такой диеты в следующем:
- Отказ от продуктов, содержащих «плохие» жиры и замена их на «хорошие», введение в рацион ненасыщенных жиров, которые содержатся в оливковом, льняном, кунжутном масле, авокадо.
- Применение в пищу мяса и рыбы, при производстве которых не были использованы антибиотики или гормоны.
- Полное удаление из рациона рафинированного сахара и глютена.
- Употребление в пищу следующих продуктов - китайская капуста, брокколи, сельдерей, ананас, лосось, свекла, кукумария, имбирь, куркума и других продуктов, обладающих противовоспалительными свойствами.
- Молочные продукты допустимы только на основе овечьего и козьего молока.
- Допустимо употребление травяных чаев, лимонада, кваса, морсов и натуральных соков.
Профилактика
Так как мышечную дистрофию довольно тяжело предсказать и обнаружить на раннем этапе, профилактические мероприятия сводятся к двум простым рекомендациям:
Обязательное обследование женщины на этапе планирования беременности на предмет наличия мутаций в генах
В случае если до беременности, по каким-либо причинам не было сделано генетического обследования, уже в период беременности проводится тест на определение у плода мутаций в Х хромосоме.
Прогноз и осложнения
В зависимости от вида болезни прогноз может быть разным, и тем не менее можно выделить несколько возможных осложнений, возникающих при данном недуге.
- нарушения сердечной деятельности
- искривление позвоночника
- снижение интеллектуальных способностей больного
- снижение двигательной активности
- нарушения дыхательной системы
- летальный итог
Итак, мышечная дистрофия довольно опасное и неизлечимое заболевание, поэтому будущим родителям необходимо с глубокой ответственностью относиться к планированию беременности. Берегите себя и своих будущих детей!
Мышечной дистрофией фактически называют группу наследственных заболеваний, характеризуемых прогрессирующей симметричной атрофией скелетных мышц, протекающей без болей и потери чувствительности в конечностях. Парадоксально, но пораженные мышцы могут увеличиваться в размерах из-за роста соединительной ткани и жировых отложений, создавая ложное впечатление крепких мышц.
До сих пор не существует средства, излечивающего мышечную дистрофию. Различают четыре основных вида этой патологии. Наиболее часто встречается мышечная дистрофия Дюшенна (50% всех случаев). Обычно заболевание начинается в раннем детстве и приводит к смерти к 20 годам. Мышечная дистрофия Беккера развивается медленнее, больные живут более 40 лет. Плече-лопаточно-лицевая и конечностно-поясная дистрофии, обычно не влияют на продолжительность жизни.
Причины возникновения
Развитие мышечной дистрофии. обусловлено различными генами. Мышечная дистрофия Дющенна и мышечная дистрофия Беккера вызываются генами, находящимися в половой хромосоме, и заболевают ими только мужчины. Плече-лопаточно-лицевая и конечностно-поясничная дистрофии не связаны с половыми хромосомами; ими заболевают и мужчины, и женщины.
Симптомы
Все разновидности мышечной дистрофии вызывают прогрессирующую атрофию мышц.
Диагностика
Врач осматривает ребенка, задает вопросы о заболеваниях у членов семьи и назначает определенные исследования. Если кто-либо из родственников болел мышечной дистрофией, врач выясняет, как протекала у него дистрофия. Анализируя полученные данные, можно прогнозировать, что ожидает ребенка. Если в семье не было больных мышечной дистрофией, электромиография позволит оценить функционирование нервов в пораженных мышцах и установить наличие мышечной дистрофии; исследование кусочка мышечной ткани () может показать клеточные изменения и наличие жировых отложений.
В медицинских центрах, оснащенных самым современным оборудованием для проведения молекулярно-биологических и иммунологических исследований, могут с точностью определить, будет ли ребенок страдать мышечной дистрофией. В этих центрах могут также провести обследование родителей и родственников на наличие у них генов, определяющих развитие мышечной дистрофии Дюшенна и мышечной дистрофии Беккера.
Виды заболевания
По степени тяжести заболевания и времени его появления различают:
Дистония Дюшенна проявляет себя в раннем возрасте (между 3 и 5 годами). Больные дети ходят вразвалку, с трудом поднимаются по лестнице, часто падают, не могут бегать. Когда они поднимают руки, лопатки у них «отстают» от туловища - этот симптом получил название «крыловидных лопаток». Обычно ребенок с мышечной дистрофией к 9-12 годам оказывается прикованным к инвалидному креслу. Прогрессирующая слабость сердечной мышцы приводит к смерти от внезапно наступившей сердечной недостаточности, дыхательной недостаточности или инфекции.
Хотя дистрофия Беккера имеет много общего с дистрофией Дюшенна, развивается она гораздо медленнее. Симптомы появляются в возрасте около 5 лет, но и после 15 лет больные дети обычно еще сохраняют способность ходить, а иногда и значительно позже.
Плече-лопаточно-лицевая дистрофия развивается медленно, течение ее относительно доброкачественное. Чаще заболевание начинается до 10 лет, но может появиться в раннем подростковом возрасте. Дети, у которых впоследствии обнаруживается эта патология, в младенческом возрасте плохо сосут; когда они становятся старше, им не удается складывать губы как для свистка, поднимать руки выше головы. У больных детей лица отличаются малоподвижностью при смехе или плаче, иногда отмечают мимику, отличную от нормальной.
Действия пациента
Если вы беспокоитесь, что у вашего ребенка, возможно, развивается мышечная дистрофия, вы можете принести фотографии или видеозаписи, которые иллюстрируют ваши конкретные проблемы. Возьмите с собой родственника или друга, который также выслушает информацию, предоставленную врачом.
Лечение
До сих пор нет средства, которое бы могло остановить прогрессирование атрофии мышц при мышечной дистрофии. Однако ортопедические устройства, а также лечебная физкультура, физиотерапия и хирургия для коррекции контрактур могут на некоторое время сохранить подвижность ребенка или подростка.
Членам семей, в которых были случаи заболевания мышечной дистрофией, следует обратиться за медико-генетической консультацией с тем, чтобы узнать, существует ли риск передачи заболевания будущему ребенку.
Осложнения
Некоторые виды мышечной дистрофии сокращают продолжительность жизни человека, часто влияют на мышцы, связанные с дыханием. Даже с помощью улучшенного механического дыхания люди, у которых мышечная дистрофия Дюшенна - наиболее распространенный тип мышечной дистрофии - как правило, умирают от дыхательной недостаточности, не дожив до 40 лет.
Многие виды мышечной дистрофии могут также снизить эффективность сердечной мышцы. Если болезнью затронуты мышцы, связанные с глотанием могут возникнуть проблемы с питанием.
Так как мышечная слабость прогрессирует, подвижность становится проблемой. Многим людям, у которых развивается мышечная дистрофия, возможно, потребуется использовать инвалидное кресло. Тем не менее, длительная неподвижность суставов, связанная с использованием инвалидной коляски, может ухудшить контрактуру, при которой конечности разворачиваются во внутреннем положении и фиксируются в нем.
Контрактуры могут также играть роль в развитии сколиоза, став причиной искривления позвоночника, что еще больше снижает эффективность работы легких у людей, которые больны мышечной дистрофией.
Профилактика
Поскольку респираторные инфекции могут стать проблемой в более поздних стадиях мышечной дистрофии, важно сделать прививку от пневмонии, а также регулярно получать прививки от гриппа.
Мышечная дистрофия, или, как ее еще называют медики, миопатия - это заболевание генетической природы. В редких случаях оно развивает по внешним причинам. Чаще всего это наследственное заболевание, для которого характерны мышечная слабость, дегенерация мышц, уменьшение диаметра скелетных мышечных волокон, а в особо тяжелых случаях - мышечных волокон внутренних органов.
Что такое мышечная дистрофия?
В процессе этого заболевания мышцы постепенно утрачивают способность к сокращению. Происходит постепенный распад. Мышечная ткань медленно, но неотвратимо заменяется жировой тканью и соединительными клетками.
Для прогрессирующей стадии характерны следующие :
- сниженный болевой порог, а в некоторых случаях практические полная невосприимчивость к болевым ощущениям;
- мышечная ткань утратила свои способности к сокращению и росту;
- при некоторых разновидностях болезни - болевые ощущения в области мышц;
- атрофия скелетных мышц;
- неправильная походка вследствие недоразвития мышц ног, дегенеративные изменения стоп вследствие неспособности выдерживать нагрузку при ходьбе;
- пациент часто желает присесть и полежать, так как у него попросту нет сил находиться на ногах - этот симптом характерен для больных женского пола;
- постоянная хроническая усталость;
- у детей - неспособность нормально учиться и усваивать новую информацию;
- изменение мышц в размерах - уменьшение в той или иной степени;
- постепенная утрата навыков у детей, дегенеративные процессы в психике у подростков.
Причины ее появления
Медицина до сих пор не может назвать все механизмы запуска мышечной дистрофии. Совершенно уверенно можно заявить одно: все причины лежат в изменении набора доминантных хромосом, которые отвечают в нашем организме за белковый и аминокислотный обмен. Без адекватного усвоения белка не будет нормального роста и функционирования мышц и костной ткани.

От вида хромосом, которые подверглись мутации, зависит течение болезни и ее форма:
- Мутация икс-хромосомы - распространенная причина мышечной дистрофии Дюшенна. Когда женщина-мать носит в себе такой поврежденный генный материал, можно говорить, что с вероятностью 70 % она передаст заболевание своим детям. При этом она зачастую патологиями мышечной и костной ткани не страдает.
- Миотоническая мышечная дистрофия проявляется по причине дефектного гена, принадлежащего девятнадцатой хромосоме.
- Половые хромосомы не влияют на локализацию мышечной недоразвитости: поясница-конечности, а также плечо-лопатка-лицо.
Диагностика заболевания
Диагностические мероприятия разнообразны. Существует множество недугов, напоминающих по тем или иным косвенными миопатии. Наследственность - самая частая причина заболевания "мышечная дистрофия". Лечение при этом возможно, но оно будет длительным и сложным. Обязательно нужно собрать сведения о режиме дня больного, об образе жизни. Как он питается, ест ли мясо и молочные продукты, употребляет ли алкогольные напитки или наркотики. Особенно важны эти сведения при диагностике мышечной дистрофии у подростков.

Подобные данные необходимы для составления плана проведения диагностических мер:
- электромиография;
- МРТ, компьютерная томография;
- биопсия мышечной ткани;
- дополнительная консультация ортопеда, хирурга, кардиолога;
- анализ крови (биохимия, общий) и мочи;
- соскоб мышечной ткани для анализа;
- генетическое тестирование для выявления наследственности пациента.
Разновидности заболевания
Исследуя на протяжении веков развитие прогрессирующей мышечной дистрофии, медики выявили следующие виды недуга:
- Дистрофия Беккера.
- Плече-лопаточно-лицевая мышечная дистрофия.
- Дистрофия Дюшенна.
- Врожденная мышечная дистрофия.
- Конечностно-поясная.
- Аутосомно-доминантная.
Это самые распространенные формы заболевания. Некоторые из них на сегодня успешно преодолимы благодаря развитию современной медицины. Некоторые имеют под собой наследственные причины, мутацию хромосом и терапии не поддаются.
Последствия недуга
Результат возникновения и прогресса миопатий различного генеза и этиологии - инвалидность. Серьезная деформация скелетных мышц и позвоночника приводит к частичной или полной утрате способности к движению.

Прогрессирующая мышечная дистрофия по мере своего развития часто приводит к развитию почечной, сердечной и дыхательной недостаточности. У детей - к умственному и физическому отставанию в развитии. У подростков - к нарушениям интеллектуальных и мыслительных способностей, остановке роста, карликовости, ухудшению памяти и потере способности к обучению.
Дистрофия Дюшенна
Это одна из самых тяжелых форм. Увы, современная медицина так и не смогла помочь больным прогрессирующей мышечной дистрофией Дюшенна адаптироваться к жизни. Большинство пациентов с этим диагнозом получают инвалидность с детства и не живут дольше тридцати лет.
Клинически проявляется уже в возрасте двух-трех лет. Дети не могут играть со сверстниками в подвижные игры, быстро утомляются. Часто наблюдается отставание в росте, в развитии речи и когнитивных функций. К пятилетнему возрасту мышечная слабость и недоразвитие скелета у ребенка становятся совершенно очевидными. Походка смотрится странно - слабые мышцы ног не позволяют больному идти ровно, не пошатываясь из стороны в сторону.
Родителям нужно начинать бить тревогу как можно раньше. Сделать как можно быстрее ряд генетических анализов, которые помогут с точностью установить диагноз. Современные методы лечения помогут больному вести приемлемый образ жизни, хотя и не восстановят полностью рост и функции мышечной ткани.
Дистрофия Беккера
Эта форма мышечной дистрофии была исследована Беккером и Кинером еще в 1955 году. В мире медицины ее именуют как мышечную дистрофию Беккера, или Беккера-Кинера.

Первичные симптомы такие же, как и у формы заболевания по Дюшенну. Причины развития так же и кроются в нарушении генного кода. Но в отличие от дистрофии Дюшенна, форма заболевания по Беккеру носит доброкачественный характер. Пациенты с этим типом болезни могут вести практически полноценную жизнедеятельность и дожить до преклонного возраста. Чем раньше диагностировать недуг и начать лечение, тем больше вероятность для пациента обычной человеческой жизни.
Не наблюдается и замедления развития умственных функций человека, свойственного злокачественной мышечной дистрофии по форме Дюшенна. При рассматриваемой болезни очень редко встречается кардиомиопатия и прочие отклонения в работе сердечно-сосудистой системы.
Плече-лопаточно-лицевая дистрофия
Эта форма заболевания достаточно медленно прогрессирует, имеет доброкачественный тип течения. Чаще всего первые проявления недуга заметны в возрасте шести-семи лет. Но иногда (примерно 15 % случаев) заболевание никак не проявляет себя до тридцати-сорока лет. В некоторых случаях (10 %) ген дистрофии не пробуждается вовсе в течение всей жизни пациента.
Как становится ясно из названия, поражаются мышцы лица, плечевого пояса и верхних конечностей. Отставание лопатки от спины и неровное положение уровня плеч, искривленная плечевая дуга - все это говорит о слабости или полной дисфункции передней зубчатой, трапециевидной и Со временем в процесс включаются двуглавые мышцы, задняя дельтовидная.

У опытного врача при взгляде на пациента может сложится обманчивое впечатление о наличии у него экзофтальма. Функция щитовидной железы при этом остается нормальной, обмен веществ чаще всего не затронут. Интеллектуальные возможности пациента также, как правило, сохранены. У больного есть все возможности вести полноценный, здоровый образ жизни. Визуально сгладить проявления плече-лопаточно-лицевой мышечной дистрофии помогут современные лекарственные препараты и физиотерапия.
Миотоническая дистрофия
Наследуется в 90 % случаев по аутосомно-доминантному типу. Поражает мышечную и костную ткань. Миотоническая дистрофия - очень редкое явление, частота его появления 1 к 10000, но эта статистика занижена, так как часто эта форма заболевания остается недиагностированной.
Дети, рожденные от матерей с миотонической дистрофией, часто страдают так называемой врожденной миотонической дистрофией. Она проявляется слабостью лицевых мышц. Параллельно часто наблюдается неонатальная дыхательная недостаточность, перебои в работе сердечно-сосудистой системы. Часто можно заметить отставание в умственном развитии, задержку психо-речевого развития у маленьких пациентов.
Врожденная мышечная дистрофия
В классических случаях гипотония заметна уже с младенческого возраста. Характерно уменьшение мышц и костной ткани в объеме наряду с контрактурами суставов рук и ног. В анализах активность сывороточного КК повышена. При биопсии пораженных мышц обнаруживается стандартная для мышечной дистрофии картина.

Эта форма носит не прогрессирующий характер, интеллект пациента почти всегда остается сохранным. Но, увы, многие больные врожденной формой мышечной дистрофии не могут передвигаться самостоятельно. Позднее может добавиться дыхательная недостаточность. Компьютерная томография иногда помогает выявить гипомиелинизацию слоев белого вещества мозга. Это не имеет известных клинических проявлений и чаще всего никак не сказывается на адекватности и психической состоятельности пациента.
Анорексия и психические расстройства как предвестники мышечного заболевания
Отказ многих подростков от приема пищи несет за собой необратимую дисфункцию мышечной ткани. Если в течение сорока дней в организм не попадают аминокислоты, не происходят процессы синтеза белковых соединений - мышечная ткань на 87 % отмирает. Поэтому родители должны следить за питанием детей, чтобы те не следовали новомодным анорексичным диетам. В рационе подростка должны ежедневно присутствовать мясо, молочные продукты и растительные источники белка.
В случаях запущенных расстройств пищевого поведения может наблюдаться полная атрофия некоторых мышечных участков, а в качестве осложнения часто появляется почечная недостаточность сначала в острой, а затем и в хронической форме.
Лечение и препараты
Дистрофия является серьезным хроническим заболеванием наследственного характера. Полностью ее вылечить невозможно, но современная медицина и фармакология позволяют откорректировать проявления недуга, чтобы сделать жизнь пациентов максимально комфортной.
Список препаратов, необходимых больным для лечения мышечной дистрофии:
- "Преднизон". Стероидное анаболическое средство, поддерживающее на высоком уровне синтез белка. При дистрофии позволяет сохранить и даже нарастить мышечный корсет. Является гормональным средством.
- "Дифенин" - также гормональный препарат со стероидным профилем. Имеет множество побочных эффектов и вызывает привыкание.
- "Оксандролон" - был разработан американскими фармацевтами специально для детей и женщин. Как и предшественники, является гормональным средством с анаболическим эффектом. Имеет минимум побочных эффектов, активно применяется для терапии в детском и юношеском возрасте.
- Гормон роста инъекционный - одно из новейших средств от мышечной атрофии и остановки роста. Очень эффективное средство, которое позволяет больным ничем не выделяться внешне. Для лучшего эффекта принимать его необходимо в детском возрасте.
- "Креатин" - натуральный и практически безопасный лекарственный препарат. Подходит детям и взрослым. Способствует росту мышц и предотвращает их атрофию, укрепляет костную ткань.
В список прогрессирующих мышечных дистрофий (ПМД) входят миодистрофии:
- псевдогипертрофическая ;
- псевдогипертрофическая Беккера-Кинера;
- Эмери-Дрейфуса-Хогана;
- Роттауфа (фиброзирующая миопатия);
- ювенильная Эрба-Рота;
- окулярная (офтальмоплегия Грефе);
- плече-лопаточно-лицевая (Ландузи);
- окулофарингеальная;
- Дрейфуса;
- митохондриальные.
Причины мышечных дистрофий
У больных ПМД выявляется врожденный структурный дефект мышечной ткани (например, при ПМД Дюшенна — дефект гена, отвечающего за синтез структурного мышечного белка дистрофина). В отличие от утраты этого белка при миодистрофии Дюшенна, при миодистрофии Беккера дистрофин качественно изменен. У больных ПМД отмечают нарушения:
- возбудимости и проводимости мышечных волокон;
- микроциркуляции;
- нейротрофических влияний;
- метаболизма мышц.
Провоцирующими факторами выступают, в частности, инфекции, интоксикации, травмы (физические и/или психические) и соматические заболевания.
Симптомы мышечных дистрофий
Общие симптомы ПМД:
- мышечная слабость (симметричная);
- отсутствие перманентной боли;
- более частое проявление слабости в проксимальных отделах, ее преобладание в мышцах тазового, плечевого пояса;
- снижение и угасание сухожильных рефлексов пропорционально мышечной слабости.
В целом клиническая характеристика семейных и спорадических форм миопатий сходна. Прогрессируют медленно и постепенно. Локализация атрофий при различных формах миопатий:
- плечевые;
- тазовые;
- тазо-плечевые;
- плече-лопаточно-лицевые;
- дистальные;
- глазные;
- глазо-бульбарные;
- смешанные.
Характер распространения мышечной дистрофии — восходящий или нисходящий. Походка принимает так называемый утиный характер. Из положения лежа больные поднимаются с помощью дополнительных движений — приемов миопата. Наряду с атрофиями наблюдаются псевдогипертрофии мышц (у 37% больных), преимущественно в икроножных и четырехглавых мышцах, реже в дельтовидных, надостных, подостных и межреберных мышцах. Слабость мышц нарастают по мере развития заболевания и приводят к снижению мышечной силы, которую в поздних стадиях оценивают в 0-1 балл (по пятибалльной системе). Одновременно с нарастанием выраженности атрофий скелетных мышц отмечается понижение и угасание сухожильных надкостничных рефлексов. У большинства больных (83,5%) определяются вегетативно-сосудистые изменения: гипергидроз, акроцианоз стоп и ладоней, повышенная лабильность вазомоторов, стойкий красный дермографизм. Для различных форм первичных миодистрофий характерны и некоторые общие симптомы и феномены.
Миодистрофия псевдогипертрофическая Дюшенна
Псевдогипертрофическая мышечная дистрофия Дюшенна — злокачественная форма ПМД. Заболевание наследуется сцепленно с Х-хромосомой. При миодистрофии Дюшенна идентифицировано генетически обусловленное отсутствие структурного мышечного белка дистрофина, что ведет к запуску каскада химических реакций, вызывающих гибель миофибрилл. В соответствии с таким типом наследования болезни болеют обычно мальчики, матери которых являются носителями рецессивного гена. При миодистрофии Дюшенна существует эффект деда: от деда заболевание может передаться через дочь внуку.
Первые признаки мышечной дистрофии появляются на первом году, а к его концу становится заметным отставание детей в развитии. С 2 лет отмечается слабость мышц.
Основные проявления
«Утиная» походка
Пациент при ходьбе «переваливается» с ноги на ногу, что обусловлено слабостью ягодичных мышц
Псевдогипертрофии — «икры гнома»
Псевдогипертрофия икр из-за их жировой инфильтрации, разрастания соединительнотканных образований. Мышцы выпуклые, плотные на ощупь, но сила снижена (признак миопатии Дюшенна)
«Лягушачий живот»
При некоторых формах нервно-мышечной патологии низкий тонус мышц приводит при мышечной дистрофии к тому, что живот выступает вперед.
Симптом «вялых надплечий»
Если ребенка приподнять, взяв его подмышки, то предплечья резко поднимаются, и голова больного «тонет» между ними — признак выраженной гипотонии мышц плечевого пояса
«Поперечная» улыбка
Слабость и гипотрофия лицевой мускулатуры могут сопровождаться изменением мимики, в частности поперечным растягиванием рта. Признак некоторых форм миопатии.
Симптом Шерешевского-Говерса
Пациент с миопатией, вставая из положения лежа, совершает ряд движений (поворачивается на живот, становится на четвереньки), а затем начинает подниматься, постепенно разгибая ноги и опираясь руками; руки его последовательно меняют положение, при этом больной «вскарабкивается» по собственным ногам, как по приставной лестнице.
Триада синдромов при мышечной дистрофии: трех «А»
- Атрофии (гипотрофии)
- Атонии (гипотонии)
- Арефлексии (гипорефлексии) тазового пояса, вследствие чего возникает «утиная» походка.
Характерное распространение при мышечной дистрофии миодистрофического процесса восходящее — со временем в процесс вовлекаются все мышцы туловища и плечевого пояса. Грудная клетка уплощена, отмечают сколиоз грудного отдела и поясничный гиперлордоз. К 7 годам больные с трудом передвигаются, к 12-15 годам они утрачивают возможность ходить.
Характерны феномены Шерешевского-Говерса, поясничный гиперлордоз, псевдогипертрофия отдельных мышечных групп. Типична псевдогипертрофия икр («икры гнома»), при подъеме больные испытывают выраженные трудности. С вовлечением мышц плечевого пояса формируются «крыловидные» лопатки, кифосколиоз, развивается слабость рук, дыхательной мускулатуры.
В поздней стадии миодистрофического процесса возникают гипотрофия мышц лица, глотки и гортани; отмечается сгибательная контрактура в суставах конечностей. Развивается кардиомиопатия (расширение границ сердца, нарушения сердечного ритма), возникают изменения электрокардиограммы (ЭКГ), возможны адипозогенитальный синдром, гипоплазия надпочечников и остеопороз.
Около трети больных (30%) отстают в развитии интеллектуальных функций. В ранней стадии резко (в десятки и сотни раз) возрастает активность КФК, а также активность ЛДГ.
Миодистрофия псевдогипертрофическая Беккера-Кинера
Псевдогипертрофическую мышечную дистрофию Беккера-Кинера рассматривают как мягкую форму ПМД Дюшенна. Заболевание также передается сцепленно с Х-хромосомой. Дебют заболевания отмечается в возрасте от 5 лет.
Течение миодистрофического процесса медленно прогрессирующее. Особенности распространения мышечных дистрофий идентичны признакам мышечной патологии при ПМД Дюшенна. Менее выражена патология сердца. Интеллект пациентов сохранен; они долго сохраняют способность самостоятельно передвигаться, могут иметь детей.
Миодистрофия Эмери
Мышечная дистрофия Эмери-Хогана наследуется сцепленно с Х-хромосомой. Рано развиваются ретракции пяточных сухожилий, при ходьбе отмечают опору на пальцы, а также проявление «утиной» походки. Отмечают миокардиодистрофию, множественные контрактуры крупных суставов, ригидность позвоночника, бочкообразную грудную клетку. Активность КФК умеренно повышена. Течение медленно прогрессирующее. Интеллект сохранен.
Миодистрофия Роттауфа (фиброзирующая)
Дебют мышечной дистрофии происходит в детском возрасте, обычно в 4-12 лет. Возникают выраженные сухожильные контрактуры. Отмечают ограничения тыльного разгибания стоп, сгибания шеи. Вследствие фиброза мышц формируются патологические позы, приводящие к невозможности сгибания позвоночника. Миодистрофический процесс медленно прогрессирует. Появляются мышечная слабость и умеренные гипотрофии, больше в лопаточно-плечевой области. Интеллект сохранен. Развивается кардиомиопатия. Характерна выраженная гиперферментемия. ЭМГ выявляет изменения, указывающие на первичный миодистрофический процесс.
Ювенильная миодистрофия
В первую очередь болезнь характеризуется атрофией мышц таза. Ранним проявлением заболевания служит «утиная» (миопатическая) походка. Пациент испытывает затруднения при попытке сесть из положения лежа. Выявляют поясничный гиперлордоз, «лягушачий» живот, атрофии верхних конечностей (форма Лейдена-Мебиуса). Возможно развитие умеренных псевдогипертрофий. На фоне вовлечения в процесс межреберных мышц, диафрагмы может возникнуть дыхательная недостаточность.
У больных вероятны эндокринопатии (ожирение) и вегетативная дистония. Течение миодистрофического процесса относительно мягкое. При неблагоприятных условиях (например, при физических нагрузках) возможно быстрое прогрессирование процесса.
На ЭМГ при мышечной дистрофии присутствует картина первичного миодистрофического процесса. На ЭНМГ скорость импульса в пределах возрастной нормы. В крови определяется умеренная гиперферментемия.
Миодистрофия плече-лопаточно-лицевая (Ландузи)
Заболевание наследуется аутосомно-доминантно. Предположительно патологический ген локализован на хромосоме 4. Отмечается выраженная пенетрантность гена.
Дебют заболевания обычно происходит к 20 годам, болезнь начинается со слабости мышц лица. Рано наблюдаются «губы тапира», улыбка Джоконды). Со временем нарастает похудание и слабость передней зубчатой и большой грудной мышц. Позже миодистрофический процесс затрагивает перонеальную группу мышц, возникает походка «степпаж». Развивается умеренная псевдогипертрофия мышц. Креатин-креатининовый обмен нарушен умеренно.
Миодистрофия окулярная (Грефе)
Заболевание дебютирует до 30-летнего возраста. Нарастает поражение глазных мышц, которое протекает без диплопии, приводит к обездвиженности глазных яблок (параличу взора). Сначала часто поражается мышца, поднимающая верхнее веко; в поздней стадии миодистрофического процесса наблюдается двусторонний птоз. Иногда вовлекаются мимические, бульбарные и скелетные мышцы.
Миодистрофия окулофарингеальная
Окулофарингеальная миодистрофия — классическая наружная офтальмоплегия с дисфагией и дисфонией, наследуемая аутосомно-доминантно. В 1961 г. заболевание описано у представителей франко-канадской популяции.
Классический вариант болезни — прогрессирующая офтальмоплегия с дисфагией и дисфонией, сопровождаемая птозом верхних век. Другой вариант болезни протекает с присоединением пареза мышц, обеспечивающих движения глаз, мимической и жевательной мускулатуры, мышц шеи. При третьем, редком варианте в процесс вовлекаются также мышцы конечностей. Активность ферментов (КФК и ЛДГ) несколько повышена.
Миодистрофия Дрейфуса
Мышечная дистрофия Дрейфуса проявляется нарастающей слабостью мышц, преимущественно тазового пояса и нижних конечностей. Характерна походка больного с опорой на большие пальцы; отмечается поясничный гиперлордоз.
Особенность этой формы миодистрофии — формирование выраженных контрактур локтевых и других суставов. Нередко поражается миокард, больной отстает в психическом развитии.
Миопатии митохондриальные
При митохондриальных миопатиях биохимический дефект локализован в митохондриях клеток, что выявляют при биохимическом и ультрамикроскопическом исследованиях. Чаще всего дебют заболевания происходит на 2-м десятилетии жизни.
На ранних стадиях возникают птоз, наружный офтальмопарез (без диплопии вследствие симметричности поражения глазодвигательных мышц), слабость проксимальных мышцах, сухожильная гипо- и арефлексия.
Длительность прогрессирования мышечной дистрофии вариабельна: от месяцев до десятилетий. Клинико-нейрофизиологические исследования (ЭМГ, ЭНМГ) при митохондральных миопатиях выявляют миодистрофические и невропатические проявления.
Диагностика
Для постановки диагноза необходимы нейрофизиологические, биохимические и патогистологические исследования.
Локальная ЭМГ. У больных миопатиями регистрируется патологическая интерференция с высокой частотой полифазных потенциалов и укорочением времени формирования отдельных осцилляций.
Биохимическое исследование. На ранних стадиях активность КФК крови увеличивается в 50 раз и более; ЛДГ — в 5-7 раз; ФДА — в 2-5 раз; в поздних стадиях снижается до возрастной нормы.
Патогистологическое исследование. Определяются признаки ПМД:
- перерождение мышечной ткани;
- беспорядочное расположение разнокалиберных мышечных волокон;
- чередование нормальных, атрофированных и (в некоторых мышцах) гипертрофированных волокон;
- атрофия мышечных волокон в длину и в поперечнике;
- пролиферация ядер с их расположением под сарколеммой и внутри волокна.
Лечение мышечных дистрофий
Лечение первичных направлено на замедление темпа развития заболевания и максимальное сохранение способности больного к самообслуживанию.
Тактика лечения ПМД:
- диетотерапия с целью избежать развития у больного избыточной массы тела;
- и двигательная активность, в частности, для поддержания функций опорных суставов и предотвращения контрактур;
- дыхательная гимнастика;
- медикаментозная терапия;
- социально-психологическая реабилитация;
- ортопедическая помощь.
При некоторых формах миодистрофий, в частности при миодистрофии Дюшенна, применение глюкокортикоидов иногда позволяет отодвигать наступление обездвиженности больного на годы. Поскольку такое лечение длительное и обычно сопровождается многими осложнениями, необходимо обдуманное назначение глюкокортикоидов. При ПМД применяют (с учетом возможных противопоказаний) преднизолон 1-2 мг/кг утром, через день, возможен и ежедневный прием преднизолона при мышечной дистрофии (утром однократно в дозе 0,75 мг/кг/сутки). Авторы предлагают применять также препарат этой же группы — дефлазакорт, обладающей меньшими побочными эффектами, приблизительно в той же дозе (по эффективности 6 мг дефлазакорта соответствуют 5 мг преднизолона). Длительность лечения зависит от эффективности и выраженности побочных действий препарата.
Показана симптоматическая и общеукрепляющая терапия. В частности, рекомендуют применение аденозина фосфата, трифосаденина, витамина Е, коферментов (например, кокарбоксилазы), негормональных анаболических средств (этилтиобензимидазола, оротовой кислоты), инозина, препаратов калия. При этом надо иметь в виду недоказанность объективных проявлений эффективности этих препаратов.
Преднизолон внутрь (утром) по 1-2 мг/кг/сутки через день, длительность терапии определяют индивидуально, или Преднизолон внутрь (утром) по 0,75 мг/кг/сутки ежедневно, длительно или Дефлазакорт внутрь по 6 мг, длительно.
Прогноз при мышечных дистрофиях
Прогноз наследственных миопатий зависит от формы. Поскольку у большинства больных заболевание носит неуклонно прогрессирующий характер, то прогноз неблагоприятный.
Больные миодистрофией псевдогипертрофической Дюшенна обычно умирают на 3-м десятилетии жизни, чаще от прогрессирующей сердечной недостаточности. При псевдогипертрофической мышечной дистрофии Беккера-Кинера и мышечной дистрофии Эмери-Дрейфуса-Хогана больные обычно доживают до 40-60 лет. При миодистрофии Роттауфа-Мортъе-Бейера пациенты нередко доживают до 30-50 лет.
Статью подготовил и отредактировал: врач-хирург